MAPbI3 구조를 기반으로 한 하이브리드 유무기 할라이드 페로브스카이트 구조의 합리적인 DFT 계산 방법 연구
Abstract
Hybrid Organic-Inorganic Perovskites (HOIP) have received a great deal of attention as a key material for applications like solar cells and light emitting devices because of their many advantages, in spite of their stability and toxicity issues. Attempting to discover and characterize novel HOIPs using just an experimental approach would be prohibitively time-and-cost-consuming. Using theoretical or empirical calculations would greatly help. For these reasons, HOIP has been actively investigated using DFT (Density Functional Theory) calculations, which have significantly reduced research time and cost. However, the input model structure treatment needs to be standardized to avoid unnecessary complications. For this purpose, a sort of optimization of DFT calculation protocols for HOIPs is essential, because DFT calculation results are greatly affected by the input model structure arrangements and exchange-correlation functionals. In this paper, we used DFT to calculate the band gap, formation energy, and effective mass of the well-known cubic perovskite structure, methylammonium lead iodide (CH3NH3PbI3: MAPbI3) with and without the van der Waals function and SOC (Spin Orbit Coupling) and various geometrical molecule arrangements in the structure. In particular, the initial orientation of the ‘A’ site molecule in the input model structure was intensively investigated in terms of band gap, formation energy and effective mass. It was found that the relaxation-induced final structure was greatly influenced by the initial orientation of the molecule and thereby significantly affected the DFT-calculated result.
Key words: hybrid organic-inorganic perovskite, density functional theory, band gap, effective mass, formation energy
1. 서 론
하이브리드 유무기 할라이드 페로브스카이트 구조(hybrid organic/inorganic perovskite: HOIP)는 높은 광 흡수 계수, 우수한 캐리어 이동도 및 저온, 저가 공정이 가능하다는 장점으로 지난 10년 동안 태양전지의 광 흡수체로 사용되면서 최근 몇 년 만에 25.5% [ 1- 4]에 이르는 매우 향상된 전력변환 효율을 보여주고 있다. 또한, 우수한 발광 효율로 발광 다이오드, 레이저, 광 검출기 등의 다양분야로도 응용이 되고 있다. 이러한 HOIP 구조는 AxByXz 화학식으로 되어 있으며, 합성 조건 및 A 자리의 유기물 종류, B 양이온, X 음이온 자리의 원소 종류 및 구조를 이루는 조성에 따라 structure type이 달라진다. 이러한 조성 및 구조 변화의 용이성으로 특성이 변화하여 많은 연구자들의 주목을 받고 있으며, 새로운 조성의 연구가 활발히 진행되고 있다 [ 5- 21]. 많은 HOIP 구조 중에 3차원 구조를 가지는 ABX 3 type 페로브스카이트 구조가 가장 많이 연구되었으며, ABX 3 화학식에서 A 양이온 자리에 대표적으로 많이 사용하는 유기물로는 메틸암모늄(MA: Methylammonium, CH 3NH 3+)이나 포름아미디늄(FA: Formamidinium, CH(NH 2) 2+), 에틸암모늄(EA: ethylammonium, CH 3CH 2NH 3+), 과니디늄(GA: guanidinium, [C(NH 2) 3]+) 등이 있으며, B 양이온 자리에는 납, 주석, 저마늄, X 음이온 자리에 요오드, 브롬 및 염소와 같은 할로겐 원소가 자리를 차지하거나 공유한다. A 자리 유기물의 경우 앞서 언급한 유기물을 제외하고도 수많은 유기물에 대한 연구가 활발히 진행되고 있으며, B 양이온 및 X 음이온 또한 연구되고 있다. 하지만, 지식과 경험을 기반으로 하는 실험적 접근법에는 많은 경우의 수를 가지는 HOIP 실험에 한계가 있다. 이러한 실험적 한계로 비용 및 시간을 절감 할 수 있는 DFT 계산 방법론이 대두되고 있으며 [ 22- 33], 많은 수의 DFT 계산 결과를 가지고 새로운 페로브스카이트 발견을 용이하게 하는 소재 머신 러닝 전략 [ 34- 37]이 주목을 받고 있다 [ 38- 40]. 하지만, HOIP의 DFT 계산은 몇 가지의 중요한 문제점을 가지고 있으며, 가장 큰 문제점은 결정 구조에서 유기물 분자의 위치를 특정 할 수 없다는 데에 있다. 이러한 유기물 분자의 경우 일부 구조가 해석되어 있지만, 대 부분은 무작위로 계산 모델을 만들어 DFT 계산을 수행한다. 이러한 무작위 계산 모델은 DFT 계산의 상이한 결과로 나오지만, 대부분의 연구자들은 이러한 문제에 대해 깊게 취급하지 않고 있다. 또 다른 문제점은 DFT 계산의 교환 상관 함수이다. DFT 계산은 교환 상관 함수에 따라 상이한 결과가 나온다. 이 문제점은 잘 알려진 사실이며, 여러 구조를 대상으로 한 많은 연구에서 보고되었다 [ 41- 43]. 이러한 맥락에서 재료 특성(전자 및 열역학)을 합리적으로 계산할 수 있는 계산 접근법이 필수적이다. 본 연구에서 우리는 비교적 간단한 MAPbI 3 HOIP 구조를 대상으로 DFT 계산을 수행하여, 다른 종류의 HOIP 구조에도 적용할 수 있는 합리적인 DFT 계산 접근법을 찾고자 연구를 수행하였다.
2. 계산 방법
DFT 계산은 Vienna ab initio simulation package (VASP5.4)[ 44- 48] 프로그램을 사용하여 수행되었으며, 공통적으로 교환 상관 에너지는 GGA-PBE(generalized gradient approximation parameterized by Perdew, Burke, and Ernzerhof) [ 44], pseudopotential로 PAW (projector-augmented-wave) potential [ 49, 50], k-mesh 샘플링에는 Monkhorst-Pack 기법을 채택하였으며, K-mesh는 2 X 2 X 2 이다. 또한, 모든 계산에서 cut-off energy는 400eV, Total energy convergence는 10 -4 eV로 수행되었으며, 개별 원자에 가해지는 힘을 0.05 eV/Å 이하로 최소화하였다.
3. 결과 및 고찰
3.1 계산 모델
합리적인 DFT 계산 접근법을 찾고자 선택한 MAPbI 3 HOIP 구조는 지금까지 합성 조건과 환경에 따라 Cubic( Pm-3m), Tetragonal(I4/ mcm), Orthorhombic( Pnma) 구조로 알려져 있다 [ 51]. 본 연구에서 DFT 계산에 사용된 초기 구조는 Pm-3m 공간 그룹 갖는 CsPbI 3 (ICSD_161481) Cubic 구조로 선택하였다. 이러한 CsPbI 3 Cubic 구조를 선택한 이유는 치환되는 유기물 분자인 MA(CH 3NH 3+) 분자 때문이다. MA(CH 3NH 3+) 분자는 일반적인 원소보다 크기 때문에 치환되는 site에 충분한 공간이 있어야 유리하기 때문이다. 또한, CsPbI 3 Cubic 구조에 MA(CH 3NH 3+) 분자를 치환하면 결정학적으로 symmetry가 깨지고, 구조는 triclinic( P1)이 된다. 이러한 상황에서 DFT 계산을 수행하면, 구조 이완 과정을 통해 완전히 symmetry는 triclinic( P1)이 된다. 따라서 초기 구조는 DFT 계산 수행에 용이 하면서 가장 간단한 구조인 Cubic 구조로 선택하였다. 선택된 CsPbI 3(ICSD_161481) Cubic 구조의 Cs 원소 자리에 서로 다른 기하학적 모양을 갖는 27개의 MA(CH 3NH 3+) 분자를 치환하여 27개의 계산 모델을 만들었다. 또한, 단위 셀 상태보다 DFT 계산의 정확성을 높이기 위해 모든 모델은 2 X 2 X 2 Super cell 상태에서 계산되었다. 전체적인 계산 모델을 만드는 과정을 그림 1에 나타내었다.
그림 1(a)를 보면 알 수 있듯이 MA(CH 3NH 3+) 분자의 서로 다른 기하학적 모양은 MA 분자의 무게 중심을 기준으로 C-N 본드가 z 축 위에 있을 때를 기준으로 x축, y축, z축을 각 축 마다 0~60도 사이에서 무작위로 설정하였다. 무작위로 설정한 MA(CH 3NH 3+) 분자는 그림 1(d)에 나타내었으며, 이렇게 생성된 MA(CH 3NH 3+) 분자를 그림 1(b)와 같이 Cs 원소 자리에 치환하였으며, 마지막으로 2 X 2 X 2 Super cell을 생성하였다. 최종적으로 27개의 계산 모델 구조를 생성하였으며, 4가지(GGA, GGA+SOC, GGA+D3, GGA+D3+SOC) 다른 상태의 DFT 계산을 수행하여 형성 에너지, 밴드 갭, 유효 질량을 조사하였다.
3.2 형성 에너지 계산
모든 소재의 불안정성은 온도, 습도, 열, 빛, 산소 등의 조건에서 야기되며, 이러한 이유로 HOIP 소재 또한 장기적 안정성에 관한 많은 연구가 진행되었다 [ 52- 59]. HOIP 소재는 습한 환경에 노출되면 급격히 분해된다. HOIP 구조의 열 안정성은 그 형성 에너지를 통해 결정된다. MAPbI 3는 열 적으로 불안정하며, 333 K 이상에서 MAI + PbI 2 화합물로 분해된다 [ 60]. 마찬가지로, 본 연구는 MAPbI 3→ MAI + PbI 2 형식의 분해를 고려하여 형성 에너지를 계산하였다. 형성 에너지는 식 (1)과 같이 계산할 수 있다. 우리는 식 (1)을 통해 기하학적으로 서로 다른 27개의 MAPbI 3 HOIP구조와 4가지(GGA, GGA+SOC, GGA +D3, GGA+D3+SOC) 다른 상태의 DFT 계산을 수행하여 형성 에너지를 계산 했다. 일반적인 DFT 계산에서 형성 에너지 계산은 음수의 값이 나오지만 HOIP 구조 같이 특정 분자가 구조에 원소 대신 치환된 특수한 상황에서 형성 에너지 계산은 종종 양수 값을 가진다. 이러한 계산에서 중요한 점은 비록 양수로 나올지라도 동일한 조건에서 나온 경향성 있는 결과는 충분한 의미가 있다.
그림 2의 GGA, GGA+SOC, GGA+D3, GGA+D3+SOC 계산 조건에서 각각의 형성 에너지 분포를 보면 계산 모델에 따라 GGA는 0.341~0.449 eV/f.u., GGA+SOC는 0.247~0.354 eV/f.u., GGA+D3는 0.417~0.507 eV/f.u., GGA+D3+SOC는 0.331~0.425 eV/f.u. 분포를 보인다. 각각의 분포 차이는 0.108, 0.107, 0.09, 0.094로 나왔다. 이러한 형성 에너지 계산 결과를 보면 알 수 있듯이 계산 모델이 형성 에너지에 많은 영향을 미치는 것을 알 수 있다. 이러한 이유는 Cs 원소 자리에 MA(CH 3NH 3+) 분자가 치환되어 구조 이완 과정에서 구조를 심각하게 왜곡시키기 때문이다. 하지만, 계산 조건에 따라서는 경향성이 일치함을 알 수 있다.
3.3 Band structure and Effective mass
MA(CH3NH3+) 분자가 CsPbI3(ABX3) Cubic 구조에서 Cs 원소가 위치한 A site에 치환되면, 엄격한 결정학적 관점에서 기존의 Cubic 대칭을 잃어버리게 된다. 따라서 모든 분자 치환 구조는 P1 대칭을 갖는 것으로 간주하는 것이 합리적이다. DFT 계산 수행에서 구조적 이완이 제약없이 완료되었을 때, 이완에 의한 왜곡으로 인해 기존의 대칭은 더욱 흐트러진다. 이러한 상황에서 밴드 구조를 구성하는 방법에는 두 가지가 있다. 첫 번째는 P1 대칭의 브릴루앙 영역으로 밴드 구조를 구성하는 것이고, 두 번째는 원래 구조 대칭의 브릴루앙 영역으로 밴드 구조를 구성하는 방법이다. 엄격한 결정학적 관점에서는 비록 P1 대칭이지만, 기존의 모든 보고에서 밴드 구조 구성을 원래의 대칭성을 사용하였기 때문에 본 연구에서도 원래의 대칭성을 사용하였다. 또한, P1 구성과 원래의 대칭에 따른 밴드 구조 구성은 많은 밴드 갭 차이로 이어지지 않지만, 서로 다른 유효 질량 값을 발생시킨다. 밴드 갭 차이보다 유효 질량의 차이가 크게 나는 이유는 특정 k 지점에서 측정한 유효 질량이 대칭 설정에 영향을 더 많이 받기 때문이다.
그림 3은 기하학적으로 서로 다른 27개의 MAPbI 3 HOIP구조와 4가지(GGA, GGA+SOC, GGA+D3, GGA+D3+SOC) 다른 조건의 DFT 계산을 수행하여 계산된 Band Gap 값을 보여준다. 계산 된 결과를 보면 크게 SOC(Spin Obrit Coupling) 상호작용을 포함한 계산과 포함하지 않는 경우로 나누어지며, 유기물인 MA(CH 3NH 3+) 분자에 의해 이완된 구조에 따라 상이한 결과를 나타내었다. 일반적인 DFT 계산에서 Band gap은 대부분 과소평가되어 좀더 정확한 계산을 위해 HSE06 계산을 통해 보정한다 [ 61- 67]. 하지만 MAPbI 3 HOIP 구조에서는 실험 광학 밴드 갭인 1.55 eV 보다 높은 과대평가의 결과가 나왔으며, SOC(Spin Obrit Coupling) 상호작용을 포함한 계산의 경우에는 과소평가 결과가 나왔다.
작고 균형 잡힌 유효 질량은 태양전지에서 전하 분리, 발광체에서 전하 재조합에 유리하기 때문에 작고 균형 잡힌 전자(m e*) 및 홀(m h*)를 필요로 한다. 여기서 m e*는 CBM(conduction band minimum) 곡률에서 도출한 유효 질량이며, m h*는 VBM(valence band maximum) 곡률에서 도출한 유효 질량이다. 그림 4는 기하학적으로 서로 다른 27개의 MAPbI 3 HOIP 구조와 4가지(GGA, GGA+SOC, GGA+D3, GGA+D3+SOC) 다른 상태의 DFT 계산에서 도출한 유효 질량을 나타낸다. 그림 4의 (a) m e*와 (b) m h*를 보면 앞서 언급한 형성에너지나 Band gap 보다 매우 많은 차이를 나타내는 것을 알 수 있다. m e* 경우 특히 GGA+SOC 계산에서 매우 큰 값의 유효 질량이 나왔으며, 그 차이는 매우 심각하게 나왔다. m h* 경우에서는 GGA+D3+SOC 계산에서 27개의 계산 모델 중에 12개의 계산 모델에서 매우 작은 값의 유효 질량 값을 보였다. 유효 질량 계산을 전체적으로 보면 DFT 계산 방법에서도 차이가 일어나지만, 그 보다 계산 모델에 따른 차이가 매우 큰 것을 알 수 있다. 이러한 이유는 구조 이완 과정에서 유기물인 MA(CH 3NH 3+) 분자가 구조를 심각하게 왜곡시켜 CBM/VBM의 곡률을 변화 시키기 때문이다. HOIP 구조의 유효 질량을 계산한 논문들을 보면 대부분 SOC 기능을 포함하여 계산하였다. 본 연구 결과에서 보듯이 일부에서 매우 작은 값으로 나오지만 계산 모델에 따라 그 차이가 심각하게 일어나는 것을 보았을 때 가장 중요한 것은 계산 모델임을 확인하였다.
4. 결 론
본 연구는 HOIP 구조의 합리적인 DFT 계산 방법을 찾고자 수행되었으며, HOIP 구조의 가장 큰 문제점인 유기물 분자의 위치를 특정 할 수 없다는 데에서 시작하였다. 우리는 HOIP 3차원 구조 중 가장 간단한 MAPbI3 HOIP 구조를 대상으로 기하학적으로 서로 다른 27개의 계산 모델을 GGA, GGA+SOC, GGA+D3, GGA+D3+SOC 4가지 조건으로 DFT 계산을 수행하여 형성 에너지, Band gap, 유효 질량(me*, mh*)을 계산하여 비교하였다. 그 결과 모든 결과에서 계산 모델에 따른 차이점을 보였으며, 계산 조건에 따라서는 비슷한 경향이 보임을 확인하였다. 하지만, 유효 질량의 경우에는 차이가 심각하게 나타났다. 이러한 모든 결과는 서로 다른 27개 계산 모델의 구조 이완 과정에서 일어났으며, 그 이유는 유기물인 MA (CH3NH3+) 분자가 구조를 심각하게 왜곡 시켰기 때문이다. 따라서 HOIP 구조의 DFT 계산에서 합리적인 형성 에너지, Band gap, 유효 질량의 계산을 위해서는 동일한 계산 조건으로 수행되어야 하며, 서로 다른 여러 개의 모델에 대해 DFT 계산을 할 필요성이 있다.
Acknowledgments
이 성과는 정부(과학기술정보통신부)의 재원으로 한국연구재단(NRF-2014R1A6A1030419, 2018R1C1B6006943)과 2020년 순천대학교 학술연구비(2020-0198) 공모과제로 연구되었음.
Fig. 1.
Schematic represent atom for the rotation of MA(CH3NH3+) molecule. 3 orange arrows represent the rotation of molecule around each x, y, z axis. (b) The representation of substituted CsPbI3 cubic structure with MA. (c) The calculated 2 X 2 X 2 supercell structure of (b). (d) The 27 MAs made from the method represented in (a) with randomly selected degrees. 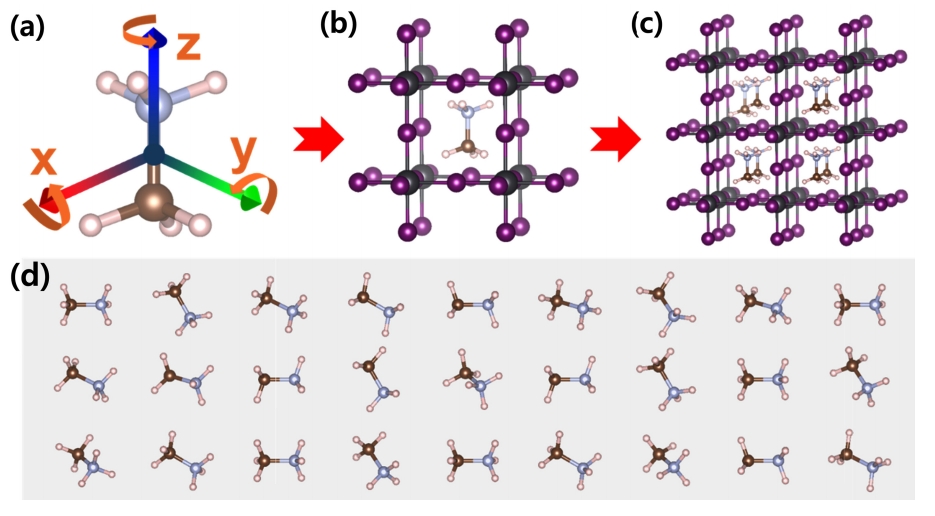
Fig. 2.
Results of formation energy calculation using different molecular configurations with four different types of DFT calculations (GGA, GGA+SOC, GGA+D3, GGA+D3+SOC). 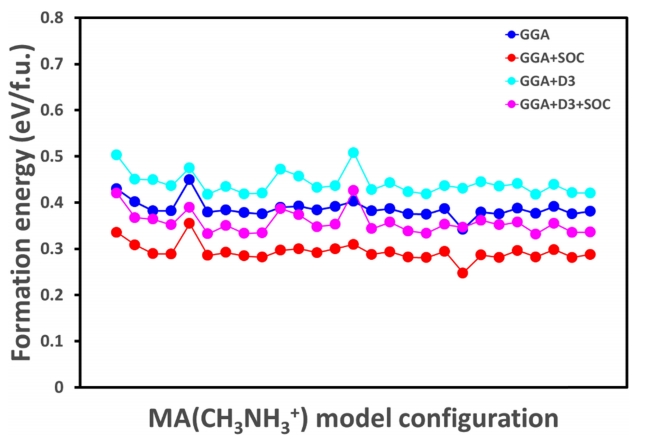
Fig. 3.
The band gap of each configurations following four different types of DFT calculations (GGA, GGA+SOC, GGA+D3, GGA+D3+SOC). 
Fig. 4.
The effective mass calculation resulted from 4 different DFT calculations (GGA, GGA+SOC, GGA+D3, GGA+D3+SOC). (a) The plot of effective mass of electron calculated from CBM (conduction band minimum) curvature. (b) The plot of effective mass of hole calculated from VBM (valence band minimum) curvature. 
REFERENCES
1. P. Basera, M. Kumar, S. Saini, and S. Bhattacharya, Phys. Rev. B. 101, 054108 (2020).  2. N. G. Park, M. Grätzel, T. Miyasaka, K. Zhu, and K. Emery, Nat. Energy. 1, 16152 (2016).  3. W. S. Yang, B. Park, E. H. Jung, and N. J. Jeon, Science. 356, 1376 (2017). 5. A. Kojima, K. Teshima, Y. Shirai, and T. Miyasaka, J. Am. Chem. Soc. 17, 6050 (2009). 6. M. M. Lee, J. Teuscher, T. Miyasaka, T. N. Murakami, and H. J. Snaith, Science. 338, 643 (2012). 7. S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza, and H. J. Snaith, Science. 342, 341 (2013). 8. M. Liu, M. B. Johnston, and H. J. Snaith, Nature. 501, 395 (2013). 9. Q. A. Akkerman, G. Rainò, M. V. Kovalenko, and L. Manna, Nat. Mater. 17, 394 (2018). 10. D.-Y. Son, S.-G. Kim, J.-Y. Seo, S.-H. Lee, H. Shin, D. Lee, and N.-G. Park, J. Am. Chem. Soc. 140, 1358 (2018). 11. A. K. Jena, A. Kulkarni, and T. Miyasaka, Chem. Rev. 119, 3036 (2019). 12. L. Mao, C. C. Stoumpos, and M. G. Kanatzidis, J. Am. Chem. Soc. 141, 1171 (2019). 13. C. Shi, L. Ye, Z.-X. Gong, J.-J. Ma, Q.-W. Wang, J.-Y. Jiang, M.-M. Hua, C. F. Wang, H. Yu, Y. Zhang, and H.-Y. Ye, J. Am. Chem. Soc. 142, 545 (2020). 14. J. Xu, X. Li, J. Xiong, C. Yuan, S. Semin, T. Rasing, and X.-H. Bu, Adv. Mater. 32, 1806736 (2019). 15. T. Nakajima and K. Sawada, J. Phys. Chem. Lett. 8, 4826 (2017). 16. Y. Li and K. Yang, Energy Environ. Sci. 12, 2233 (2019). 17. S. Lu, Q. Zhou, Y. Ouyang, Y. Guo, Q. Li, and J. Wang, Nat. Commun. 9, 3405 (2018). 18. Z. Li, Q. Xu, Q. Sun, Z. Hou, and W.-J. Yin, Adv. Funct. Mater. 29, 1807280 (2019). 19. J. Schmidt, J. Shi, P. Borlido, L. Chen, S. Botti, and M. A. L. Marques, Chem. Mater. 29, 5090 (2017). 20. J. Jeong, D. Song, J. Choe, C.-H. Chung, and K.-H. Hong, Korean J. Met. Mater. 57, 535 (2019). 21. B. T. Tan, J. Zhang, K. V. Sopiha, and P. Wu, Met. Mater. Int. 25, 869 (2019). 22. M. Kim, S. P. Singh, S. Shim, W. B. Park, and K.-S. Sohn, Chem. Mater. 32, 6697 (2020). 23. W. B. Park, M. G. T. Nathan, S. C. Han, J.-W. Lee, K.-S. Sohn, and M. Pyo, RSC Adv. 10, 43273 (2020). 24. S. C. Han, W. B. Park, K.-S. Sohn, and M. Pyo, Inorg. Chem. Front. 7, 2023 (2020). 25. J. Hong, J. Lee, Y.-W. Lee, W. B. Park, D. Ahn, J. B. Park, S. Pak, J. Baik, S. M. Morris, S. Cha, K.-S. Sohn, and J. I. Sohn, J. Power Sources. 444, 227301 (2019). 26. S. J. Richard Prabakar, A. B. Ikhe, W. B. Park, K.-C. Chung, H. Park, K.-J. Kim, D. Ahn, J. S. Kwak, K.-S. Sohn, and M. Pyo, Adv. Sci. 6, 1902129 (2019). 27. S. C. Han, W. B. Park, K.-S. Sohn, and M. Pyo, J. Solid State Electr. 23, 3135 (2019). 28. N. Naveen, W. B. Park, S. P. Singh, S. C. Han, D. Ahn, K.-S. Sohn, and M. Pyo, Small. 14, 1803495 (2018). 29. W. B. Park, S. C. Han, C. Park, S. U. Hong, U. Han, S. P. Singh, Y. H. Jung, D. Ahn, K.-S. Sohn, and M. Pyo, Adv. Energy Mater. 8, 1703099 (2018). 30. N. Naveen, W. B. Park, S. C. Han, S. P. Singh, Y. H. Jung, D. Ahn, K.-S. Sohn, and M. Pyo, Chem. Mater. 30, 2049 (2018). 31. J.-W. Lee, S. P. Singh, M. Kim, S. U. Hong, W. B. Park, and K.-S. Sohn, Inorg. Chem. 56, 9814 (2017). 32. Y. H. Jung, W. B. Park, M. Pyo, K.-S. Sohn, and D. Ahn, J. Mater. Chem. A. 5, 8939 (2017). 33. B. D. Lee, W. B. Park, J.-W. Lee, M. Kim, M. Pyo, and K.-S. Sohn, Chem. Mater. 33, 782 (2021). 34. J.-W. Lee, W. B. Park, J. H. Lee, S. P. Singh, and K.-S. Sohn, Nat. Commun. 11, 1 (2020).
35. J.-W. Lee, W. B. Park, B. D. Lee, S. Kim, N. H. Goo, and K.-S. Sohn, Sci. Rep. 10, 1 (2020). 36. K.-S. Sohn, J. Chung, M.-Y. Cho, S. Timilsina, W. B. Park, M. Pyo, N. Shin, K. Sohn, and J. S. Kim, Sci. Rep. 7, 1 (2017). 37. W. B. Park, J. Chung, J. Jung, K. Sohn, S. P. Singh, M. Pyo, N. Shin, and K.-S. Sohn, IUCrJ. 4, 486 (2017). 38. M. L. Agiorgousis, Y.-Y. Sun, D.-H. Choe, D. West, and S. Zhang, Adv. Theory Simul. 2, 1800173 (2019). 39. P. V. Balachandran, A. A. Emery, J. E. Gubernatis, T. Lookman, C. Wolverton, and A. Zunger, Phys. Rev. Mater. 2, 043802 (2018). 40. C. J. Bartel, C. Sutton, B. R. Goldsmith, R. Ouyang, C. B. Musgrave, L. M. Ghiringhelli, and M. Scheffler, Sci. Adv. 5, eaav0693 (2019). 41. W. G. Han, W. B. Park, S. P. Singh, M. Pyo, and K.-S. Sohn, Phys. Chem. Chem. Phys. 20, 26405 (2018). 42. S. U. Hong, S. P. Singh, M. Pyo, W. B. Park, and K.-S. Sohn, Phys. Chem. Chem. Phys. 19, 16702 (2017). 43. W. B. Park, S. U. Hong, S. P. Singh, M. Pyo, and K.-S. Sohn, ACS omega. 1, 483 (2016). 44. J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996). 45. G. Kresse and J. Hafner, Phys. Rev. B. 47, 558 (1993). 46. G. Kresse and J. Hafner, Phys. Rev. B. 49, 14251 (1994). 47. G. Kresse and J. Furthmüller, Comput. Mater. Sci. 6, 15 (1996). 48. G. Kresse and J. Furthmüller, Phys. Rev. B. 54, 11169 (1996). 49. P. E. Blöchl, Phys. Rev. B. 50, 17953 (1994). 50. G. Kresse and D. Joubert, Phys. Rev. B. 59, 1758 (1999). 51. P. S. Whitfield, N. Herron, W. E. Guise, K. Page, Y. Q. Cheng, I. Milas, and M. K. Crawford, Sci. Rep. 6, 35685 (2016). 52. P. Docampo and T. Bein, Acc. Chem. Res. 49, 339 (2016). 53. Y. Rong, L. Liu, A. Mei, X. Li, and H. Han, Adv. Energy Mater. 5, 1501066 (2015). 54. T. Leijtens, G. E. Eperon, N. K. Noel, S. N. Habisreutinger, A. Petrozza, and H. J. Snaith, Adv. Energy Mater. 5, 1500963 (2015). 55. T. A. Berhe, W.-N. Su, C.-H. Chen, C.-J. Pan, J.-H. Cheng, H.-M. Chen, M.-C. Tsai, L.-Y. Chen, A. A. Dubale, and B.-J. Hwang, Energy Environ. Sci. 9, 323 (2016). 56. M. Ye, X. Hong, F. Zhang, and X. Liu, J. Mater. Chem. A. 4, 6755 (2016). 57. N. H. Tiep, Z. Ku, and H. J. Fan, Adv. Energy Mater. 6, 1501420 (2016). 58. F. Brivio, J. M. Frost, J. M. Skelton, A. J. Jackson, O. J. Weber, M. T. Weller, A. R. Goñi, A. M. A. Leguy, P. R. F. Barnes, and A. Walsh, Phys. Rev. B. 92, 144308 (2015). 59. B. Li, Y. Li, C. Zheng, D. Gao, and W. Huang, RSC Adv. 6, 38079 (2016). 60. Q. Wang, Q. Dong, T. Li, A. Gruverman, and J. Huang, Adv. Mater. 28, 6734 (2016). 61. J. H. Lee, S. P. Singh, M. Kim, M. Pyo, W. B. Park, and K.-S. Sohn, Inorg. Chem. Front. 6, 3493 (2019). 62. M. Kim, W. B. Park, J.-W. Lee, J. Lee, C. H. Kim, S. P. Singh, and K.-S. Sohn, Chem. Mater. 30, 6936 (2018). 63. S. P. Singh, M. Kim, W. B. Park, J.-W. Lee, and K.-S. Sohn, Inorg. Chem. 55, 10310 (2016). 64. W. B. Park, H. Kim, H. Park, C. Yoon, and K.-S Sohn, Inorg. Chem. 55, 2534 (2016). 65. M. Kim, W. B. Park, B. Bang, C. H. Kim, and K.-S. Sohn, J. Mater. Chem. C. 4, 1914 (2016). 66. M. Kim, W. B. Park, B. Bang, C. H. Kim, and K.-S. Sohn, J. Mater. Chem. C. 3, 5484 (2015). 67. W. B. Park, S. P. Singh, M. Kim, and K.-S. Sohn, Inorg. Chem. 54, 1829 (2015).
|
|











