Zn-ion 배터리용 ZnMn2O4 양극재의 입자 크기 증가를 통한 Mn3+ 용출 완화로 수명 증가
Alleviating Mn3+ Dissolution in ZnMn2O4 Cathode for the Extended Cyclability via Particle Size Increase
Article information
Trans Abstract
ZnMn2O4 (ZMO) cathode possesses a high theoretical capacity of 224 mAh g-1 and high operating voltage (1.9 V vs. Zn2+/Zn) for aqueous Zn–ion batteries. However, the disproportionation reaction of Mn3+ leads to Mn dissolution in the ZMO cathode, deteriorating lifespan. In this study, we attempted to reduce Mn dissolution by enlarging the particle size, thereby diminishing the electrode/electrolyte interfacial area. The ZMO particle grew with increasing the calcination temperatures of 400℃, 500℃, and 600℃. Higher calcination temperature created oxygen vacancies within the lattice, thereby increasing the contents of Mn3+ for charge neutrality. The rate capability decreased with the increase in particle size, which is presumed to be due to the lengthening of the diffusion path of Zn ions. After a long–cycle experiment of Zn–ion batteries assembled with ZMO cathode and Zn anode, the Mn deposit amount on the anode was measured to reveal the Mn dissolution from the ZMO cathode based on the disproportionation reaction. The ZMO particle synthesized at 600℃ with the largest particle size demonstrated the highest cyclability of 48.1% at 1.0 A g-1 based on the lowest Mn deposit on the anode. Hence, the ZMO electrode with a larger particle size exhibited improved cycle stability by alleviating of the disproportionation reaction from the reduced electrode/electrolyte interfacial area.
1. 서 론
수계 Zn 이온 배터리의 양극소재로 Mn 기반의 소재는 1.9 V의 높은 작동 전압과 200 mAh g–1 이상의 높은 이론 용량으로 활발히 연구되고 있다[1-4]. 더욱이, 지각 내 Mn의 풍부한 매장량과 저렴한 가격으로 상업적 가치가 높다[5,6]. Mn 기반의 전극 물질 중 ZnMn2O4(ZMO)는 3차원 스피넬(spinel) 구조(공간군: I41/amd)를 가지며 결정 구조내 Zn와 Mn은 각각 사면체 자리(4a)와 팔면체 자리 (8d)에 위치한다[7,8]. 게다가 ZMO는 224 mAh g–1의 높은 이론 용량을 가지며 3차원 이온 이동이 가능하다는 장점을 갖는다[9,10]. 한편, ZMO 구조 내 Mn은 충방전 시 Jahn−Teller 효과로 인한 구조왜곡 발생과 불균등화 반응으로 인한 Mn 용출은 소재의 전기화학적 성능이 저하시킨다[11-15]. ZMO의 불균등화 반응은 전극/전해질 계면에서 주요하게 일어나며 Mn3+가 불안정한 오비탈 구조로 인해 Mn2+와 Mn4+로 산화수가 변화하면서 시작된다[13-15]. Mn2+는 이온 상태로 전해질 내 용출이 되고, Mn4+는 전극 표면에서 MnO2(s)의 새로운 화합물을 형성한다 (식 (1)) [15-17].
ZMO 전극을 Zn 이온 배터리의 양극소재로 사용하면 양극에서 용출된 Mn2+는 셀 내에서 음극으로 이동하여 증착되어 셀의 저항을 증가시킨다. 게다가, Mn의 지속적인 용출은 전하 저장 반응에 참가하는 물질의 감소를 의미하므로 전체 셀의 용량 감소를 일으킨다[18]. 따라서 Mn의 불균등화 반응의 감소를 통해 Zn배터리의 전기화학적 수명을 향상시키는 것이 ZMO 전극을 양극소재로 활용하는데 중요한 과제이다. ZnMn2O4와 유사한 구조와 특성을 가지는 Li 이온 배터리 양극소재 LiMn2O4(LMO)에 대해서 Mn 용출을 완화시키는 다양한 연구가 이루어졌다[19-22]. Lee et al.는 LiMn2O4 전극에서 Mn 자리에 용출되지 않는 원소인 Al3+을 도핑하여 수명을 향상시켰다[23]. Xiong et al.는 동일한 전극소재의 Mn 자리에 Ti4+를 도핑하여 수명 향상을 유도하였다[24]. Lu et al.은 LiMn2O4 양극소재 입자에 나노 단위의 표면 코팅을 통해 Mn 용출을 완화하여 수명 안정성 향상을 보고하였다[25]. Z hu et al.은 LMO 합성과정에서 용매인 에탄올과 물의 비율을 통해 입자크기를 증가시켜 Mn 용출을 완화시켰고 수명 안정성을 향상시켰다[26]. 즉, 양극소재의 Mn 용출 완화는 셀의 수명 안정성을 효과적으로 향상시켰다.
본 연구에서는 Zn 이온 배터리의 양극 소재인 ZnMn2O4의 입자 크기를 증가시켜 Mn 용출 반응의 억제로 수명 안정성을 향상시키고자 하였다. ZMO 내 Mn의 용출 반응은 전극/전해질 계면에서 일어나므로 입자 크기의 증가는 Mn 용출을 완화시킬 수 있을 것이다. 따라서, 전극/전해질 계면의 면적 감소를 위해 ZMO의 합성 단계에서 하소 온도를 증가시켜 큰 입자를 갖는 ZMO를 합성하고자 하였다. 또한, ZMO의 입자 크기가 물리적, 전기화학적 특성에 미치는 영향에 대해서 분석하였다.
2. 실험 방법
2.1 재료 합성
ZnMn2O4 전극은 졸–겔 법을 통해 합성되었다 (그림 1). Zinc acetate dihydrate (99.0%, Samchun, Republic of Korea), manganese acetate anhydrous (98+%, Alfa Aesar, USA)와 citric acid anhydrous (99.5%, Duksan, Republic of Korea)를 각각 1 mmol(0.2195g), 2 mmol (0.3461 g) 그리고 3 mmol(0.5764 g)의 비율로 준비하였고 citric acid 는 킬레이트제(chelating agent)로서 활용되었다. 준비된 전구체들은 초순수 증류수 (30mL)가 담겨 있는 이중 자켓 비커에서 80°C 온도로 2 시간 동안 교반하여 Zn–Mn citrate 졸을 형성시켰다. 교반이 완료된 졸 수용액은 대류 오븐(LO−FS050, Shinmyung Science, Republic of Korea)에서 80 °C에서 16 시간 동안 용매를 증발시켜 Zn–Mn citrate 겔을 제조하였다. Zn–Mn citrate 겔은 수평형 튜브 전기로를 사용하여 대기 분위기로 다양한 하소 온도 조건(400 °C, 500 °C, 600 °C)에서 4시간 동안 하소하여 ZMO를 합성하였다. 준비된 샘플은 400 °C, 500 °C, 600 °C 하소 온도에 따라 ZMO400, ZMO500 그리고 ZMO600으로 명명하였다.

Scheme of synthesis procedures for ZnMn2O4 powder through sol–gel and calcination.
2.2 재료 특성
합성된 ZMO의 표면 형상은 field−emission scanning electron microscopy (FE−SEM, SU8220, Hitachi, Japan) 를 이용하여 5 kV의 가속전압과 11.4−11.7 mm의 작동거리에서 관측하였다. 합성된 입자들의 평균 직경은 FE–SEM 이미지로부터 Image J 소프트웨어를 활용하여 구형상당지름법(equivalent spherical diameters for nonspherical particles)으로 계산하였다. 합성된 샘플들의 비표면적, 평균 기공 크기는 고진공 가스 흡착 분석기(Autosorb iQ−XR/AG, Quantachrome Instruments, USA)를 사용하여 분석하였다. 비표면적은 질소 등온곡선으로부터 multi–point Brunauer–Emmett–Teller (BET) 이론으로 계산하였으며 평균 기공 크기와 기공분포는 quenched solid density functional theory (QSDFT) 방법으로 측정하였다. 격자 구조 분석을 위하여 샘플에 대해 X–ray diffractometer (XRD, EMPYREAN, Malvern Panalytical, UK)를 활용하여, 0 − 90 °(2θ) 범위에서 40 kV, 30 mA 조건의 Cu–Kα선(λ = 1.54 Å)으로 측정하였다. Fourier transform infrared spectroscopy (FT–IR, Frontier, PerkinElmer, USA)를 활용하여 400–4000 cm–1의 파장 범위에서 Zn–O와 Mn–O 결합 특성과 Mn의 산화수 변화를 분석하였다. X–ray photoelectron spectroscopy (XPS, NEXSA, Thermo Fisher, USA)는 Al–Kα (1486.6 eV)의 방사선원을 활용하여 다른 하소온도에서 합성된 ZMO 샘플들의 소재내 Mn의 평균 산화수를 분석하였다. 또한, C 1s의 binding energy인 284.6 eV로 calibration하였고, CasaXPS 소프트웨어를 통해 Gaussian (70%)–Lorentzian (30%) function을 이용하여 fitting하였다. 홀 효과 측정기 (HMS–3000, Ecopia, Republic of Korea)를 사용하여 ZMO 펠렛의 전기전도도를 측정하였다. 펠렛은 앞서 합성한 ZMO, Ketjen black (EC 600JD, Lion Corporation, Japan), 그리고 polytetrafluoroethylene (Sigma–Aldrich, USA)를 8:1:1의 질량비로 섞어 0.2 mm 두께와 10 mm 직경의 원판형으로 제작하였다. 음극에 전착된 Mn 함량은 Inductively coupled plasma (ICP; Optima 7300DV, PerkinElmer, USA)를 활용하여 분석하였다.
2.3 전극 제작 및 전기화학 분석
합성한 ZMO 샘플, Ketjen black 도전재 그리고 polyvinylidene fluoride (Sigma Aldrich, USA) 바인더를 질량비 8:1:1로 혼합 후, 1−methyl−2−pyrrolidone (99%, Alfa Aesar, USA) 용매를 활용하여 슬러리 형태로 전극물질을 준비하였다. 전극물질을 carbon paper (HCP030N, WizMAC, Republic of Korea) 집전체에 닥터 블레이드법을 통해 1.3–2.0 mg의 질량으로 코팅한 후, 진공오븐 (OV−11, JEIO TECH, Republic of Korea)을 활용하여 80°C에서 12 h 동안 건조하였다.
앞서 제작된 전극은 양극(직경=10 mm)으로, 분리막으로 는 유리섬유여과지 (GF6 grade, CHMLAB Group, Spain)를, 그리고 음극으로는 Zn foil (99.2%, The Nilaco corporation, Japan)를 활용하여 CR2032 type의 코인셀을 제작하였다. 또한, 전해질로 활용된 2 M ZnSO4 + 0.1 M MnSO4는 zinc sulfate heptahydrate (99.0%, Duksan, Republic of Korea)와 manganese sulfate monohydrate (99%, Alfa Aesar, USA)을 초순수 증류수에 용해시켜 제작하였다. 제작된 코인셀은 상온에서 12 h 동안 시효 (aging) 후, 전기화학 측정을 진행하였다.
Electrochemical impedance spectroscopy (EIS)과 cyclic voltammetry (CV)은 potentiostat (VMP3, Bio Logic Science Instruments, France)를 사용하여 분석하였다. EIS 는 개방회로전압에서 10 mV의 진폭의 사인 파형을 중첩하여 1000kHz – 100mHz 주파수 범위에서 측정되었다. CV는 0.1mV s–1의 주사속도로 1.0–1.9 V(Zn2+/Zn)의 전압범위에서 측정하였다. 또한, Galvanostatic charge–discharge (GCD) 분석을 통하여 1.0–1.9 V(Zn2+/Zn)의 전압 범위에서 비용량, 율속 특성 및 수명 특성을 평가하였다. 비용량은 0.1 A g–1의 전류밀도에서, 율속 특성은 0.1–2.0 A g–1의 전류밀도 범위에서 2.0 A g–1/0.1 A g–1의 비용량 비율로 측정하였다. 수명 특성은 1.0 A g–1의 전류밀도에서 800회 충방전동안 용량 유지율을 통해 계산하였다. 모든 전극들의 정전류 충방전 실험 시 초기의 30 cycles은 활성화 (activation) 과정으로 간주하였으며 측정은 Battery cycler (WBCS–3000, WonA Tech, Republic of Korea) 를 사용하였다.
3. 결과 및 고찰
하소 온도를 달리하여 Zn–Mn citrate로부터 합성된 ZMO 샘플들의 형상 변화를 FE–SEM을 통하여 관찰하였다 (그림 2(a-c)). 또한, ZMO 샘플들의 입자크기는 FE–SEM 결과로부터 Image J 소프트웨어를 활용하여 측정하였다. 낮은 하소 온도에서 합성된 ZMO400, ZMO500 샘플은 미세하고 둥근 형태의 입자가 관측되었고, 높은 온도에서 제조된 ZMO600 샘플은 불균일한 형상을 보였다. 또한, ZMO400, ZMO500 그리고 ZMO600의 입자 크기는 각각 12.2, 41.8 그리고 93.7 nm였다 (표 1). 높은 하소 온도는 핵 생성 후 인접한 ZnMn2O4 입자들간 오스트발트숙성(ostwald ripening), 합체(coalescene) 등이 일어날 때 물질 확산 속도를 증가시켜 ZMO 입자의 성장 속도를 가속화한다[27-29]. 따라서, ZMO 샘플들 또한 높은 온도에서 하소할수록 입자 성장 속도가 가속화되어 평균 입자 크기가 증가한 것으로 생각된다.
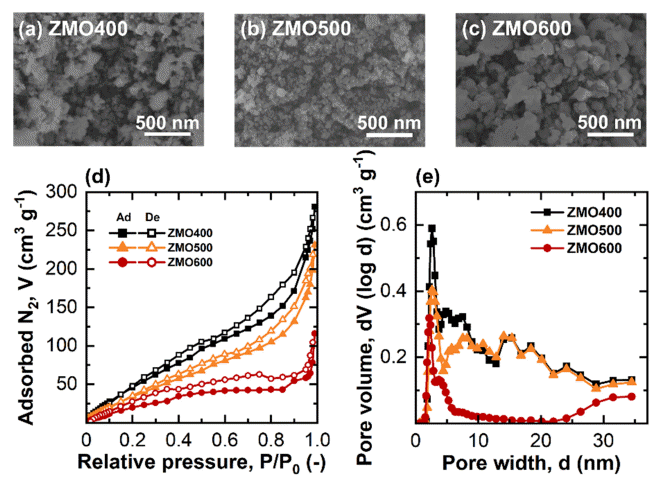
Field-emission scanning electron microscopy images of (a) ZMO400, (b) ZMO500, and (c) ZMO600. (d) N2 adsorption/desorption isotherm curves and (e) Pore size distributions of the ZMO samples at different calcination temperatures.
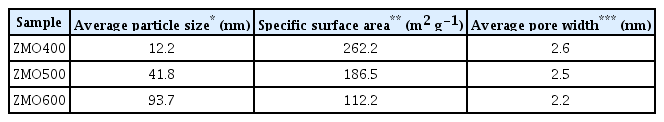
The average particle size, specific surface area, and average pore width of the ZMO samples at different calcination temperatures.
ZMO 샘플들의 입자 크기 및 비표면적, 기공 분포는 N2 흡착/탈착 분석법으로 평가하였다 (그림 2(d,e)). 하소 온도가 400 °C에서 600 °C로 높아질수록 비표면적은 각각 262.2에서 112.2 m2 g–1으로 감소하였다 (표 1). 비표면적의 감소는 하소 온도 증가에 따른 입자의 성장에 따른 것으로 생각된다. ZMO 샘플들에 대해 N2 흡탈착의 등온 곡선에서 나타난 이력 곡선(hysteresis loop)은 합성된 모든 ZMO에서 mesopore(2 ≤ d ≤ 50 nm)가 형성된 것을 의미한다 (그림 2(d)) [30]. 또한, IUPAC의 이력곡선 분류(H1 – H4)에 의하면 ZMO400, ZMO500 샘플의 경우 이력 곡선이 slit 형태의 기공을 가지는 type H3에 가까우며 높은 온도에서 하소한 ZMO600의 이력 곡선은 type H3보다 type H4에 더 가깝게 나타난다[31,32]. 이러한 이력 곡선 개형은 더 좁은 형태의 slit 기공이 높은 하소 온도 (600°C)에서 생성되었다는 것을 나타낸다[31,33,34]. 높은 하소 온도에서는 ZMO 입자 크기의 증가로 인하여 입자간 간격이 좁아져 입자 사이 기공이 좁아진 것으로 생각된다. 따라서, ZMO 샘플들의 평균 기공 크기를 측정한 결과, 입자 크기 증가에 따라 기공 크기가 감소한 것을 확인하였다 (그림 2(e), 표 1).
Zn–Mn citrate의 하소 온도를 달리하여 합성한 ZMO 샘플들의 결정 구조 변화는 X–ray 회절 패턴을 통해 분석하였다 (그림 3). ZMO400, ZMO500 그리고 ZMO600 샘플 모두 스피넬 구조의 ZnMn2O4가 합성되었다 (그림 3(a)). 높은 온도에서 하소한 ZMO 샘플들은 (211) 피크 세기가 증가하였고 반치폭(FWHM)은 감소하였으며 이는 결정성이 향상되었음을 보여준다 (표 2) [35, 36]. ZMO의 합성 과정에서 높은 하소 온도는 결정의 성장을 촉진시키고 불규칙적인 결정 구조를 규칙적이고 주기적인 결정 구조로 배열시켜 결정성을 향상시킨다[37-39]. 따라서, 높은 온도에서 하소한 ZMO600은 가장 우수한 결정성을 보이고 낮은 온도에서 하소된 ZMO 샘플은 낮은 결정성을 보인 것으로 생각된다[36].

(a) X–ray diffraction patterns of ZMO samples synthesized at different calcination temperatures (b) crystal structure of ZnMn2O4.
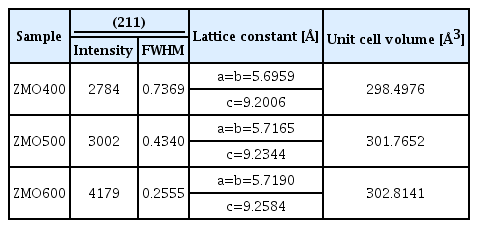
(211) peak intensity and full width at half maximum (FWHM) of X–ray diffraction pattern. Lattice constant and unit cell volume calculated via Rietveld refinement for each sample.
높은 하소 온도에서 가해진 높은 열에너지는 ZMO의 Mn–O 결합을 끊고 Mn 팔면체의 산소 자리에 공공 (vacancy)을 생성할 수 있다[40-42]. 격자 내 산소 함량이 감소하면, 전하 중성을 위해 ZMO 내 Mn4+(0.53 Å) 양은 감소하고 Mn3+(0.58 Å) 양이 증가할 것으로 예상된다[43,44]. 더 큰 이온 반경을 가지는 Mn3+의 증가는 ZMO의 격자 상수(lattice constant)와 단위격자 부피(unit cell volume)를 증가시킬 것이다[45]. Rietveld refinement 로 ZMO400, ZMO500 그리고 ZMO600 분석한 결과, 하소 온도 증가에 따라 격자상수와 단위격자 부피가 증가하는 것이 확인되었다 (그림 4, 표 2). 즉, 하소 온도 증 가에 따라 ZMO 내 산소 함량이 감소하여 Mn3+가 증가한 것으로 생각된다.

Rietveld refinement results of (a) ZMO400, (b) ZMO500 and (c) ZMO600.
Zn–Mn citrate의 하소 온도 변화에 따른 ZMO 내 Mn의 산화수 변화를 분석하기 위하여 FT–IR 분석을 수행하였다 (그림 5). FT–IR spectra에서 높은 파수(wave number)의 결합은 낮은 파수를 갖는 결합보다 짧은 결합 길이를 갖는다[46,47]. 모든 준비된 샘플에서 관측된 485 cm–1에서의 피크는 Mn–O 결합을 나타낸다. 609 cm–1 에서 관측된 피크는 Mn–O에 비해 결합길이가 짧은 Zn–O 결합의 진동에 해당하는 피크이다[46]. 또한, ZMO 합성시 하소 온도가 높아질수록 544 와 666 cm–1에서 관측되는 각각 Mn–O 와 Zn–O 결합에 해당하는 숄더 피크 (shoulder peak)의 세기가 증가하였다. 숄더 피크 세기의 증가 원인은 하소 온도 증가에 따라 ZMO 구조 내 [MnO6] 팔면체 혹은 [ZnO4] 사면체에서 산소 공공이 생성되어 Mn–O와 Zn–O의 결합 길이가 감소했기 때문으로 추측된다[44,48-50]. 즉, 산소 공공 생성으로 짧아진 Mn–O와 Zn–O 결합의 증가로 인해 숄더 피크 세기가 증 가한 것으로 생각된다[48,49].

(a) Fourier–transform infrared spectra (FT–IR) of ZMO samples at different calcination temperature. (b) Inset of FT–IR spectra indicating transition metal–oxygen bonds.
앞선 XRD와 FT–IR 결과에서 하소 온도 증가에 따라 소재내 Mn3+/Mn4+의 비율이 증가한 것으로 판단된다 (그림 3, 그림 5). ZMO 샘플들의 Mn 평균 산화수 변화를 정량적으로 분석하기 위하여 X–ray photoelectron spectroscopy(XPS)를 활용하였다 (그림 6). Mn 2p의 high resolution 스펙트럼에서 Mn3+와 Mn4+에 해당하는 피크 면적 비로부터 각 산화수의 함량을 계산하였다 (그림 6(a–c)). 또한, 다른 산화수의 Mn 함량으로부터 계산된 평균 산화수는 ZMO400, ZMO500, ZMO600에 대해 각각 3.7, 3.6, 그리고 3.4 였다 (표 3). 높은 하소 온도는 [MnO6] 팔면체 내 산소를 방출시켜 산소 공공을 생성한다[40-42]. O 1s spectra에서 산소 공공에 해당되는 영역 (Ov)과 산소 격자자리에 산소가 존재하는 경우에 해당하는 영역(OL)의 비율로부터 산소 공공의 비율을 간접적으로 확인 가능하다 (그림 6(d–f)) [51,52]. ZMO400, ZMO500, 그리고 ZMO600 샘플에서 산소 공공에 해당하는 영역은 각각 20.0, 20.9 그리고 32.3%로, 하소 온도 증가에 따라 산소 공공의 비율 증가가 확인된다 (표 3). 따라서, 하소 온도 증가에 따라 ZMO내 Mn 평균 산화수가 감소하는 이유는 격자구조 내 산소 공공이 증가하여 Mn3+의 비율이 증가했기 때문으로 생각된다[44].
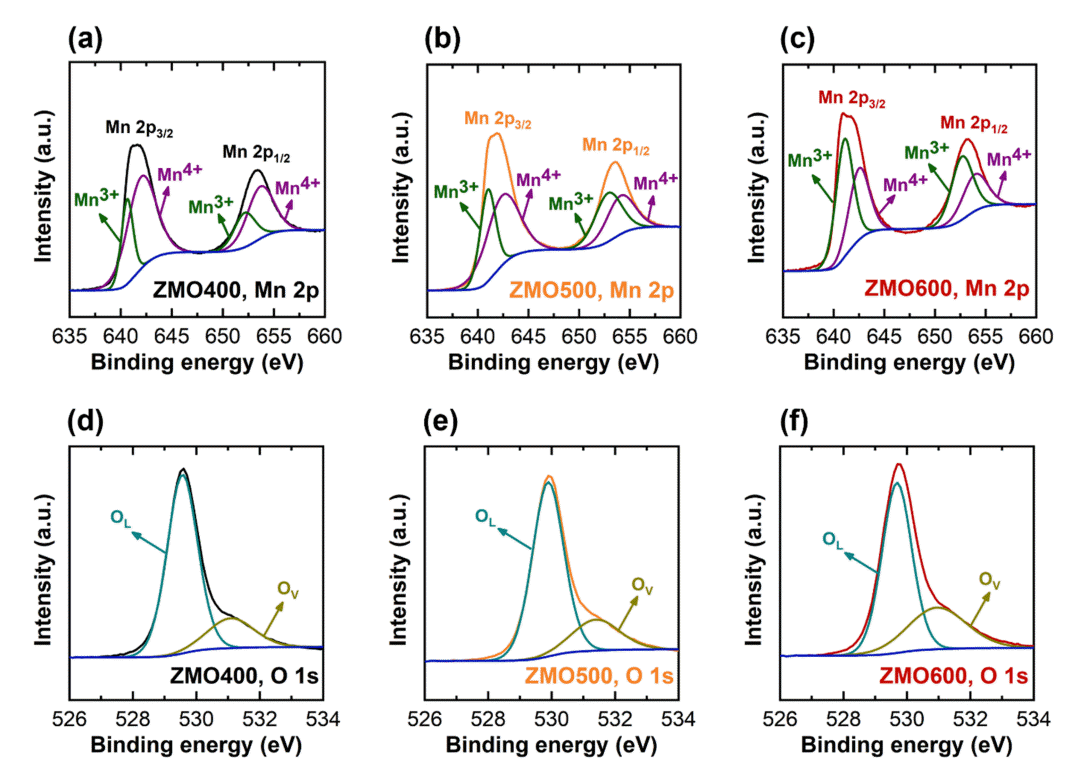
(a) X–ray photoelectron spectra of Mn 2p for (a)ZMO400, (b)ZMO500, and (c)ZMO600. O 1s spectra of (d) ZMO400, (e) ZMO500, and (f) ZMO600.
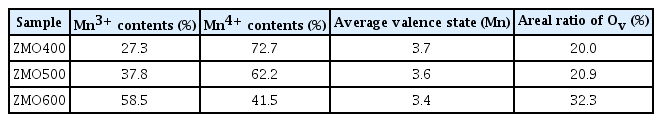
The percentage of Mn3+ and Mn4+ contents, average valence state of Mn, and the peak areal ratio of oxygen vacancy (Ov) in ZMO samples.
다른 하소 온도에서 합성된 ZMO 샘플들의 전기화학적 거동 차이는 cyclic voltammetry를 활용하여 분석하였다 (그림 7(a)). 1.55, 1.60 V에서 관측되는 2개의 산화 피크와 1.22, 1.39 V에서 관측되는 2개의 환원 피크는 Mn3+/4+의 산화·환원 반응과 관련된 피크이다[53]. 또한, ZMO 샘플들의 전기화학적 성능은 0.1 A g–1의 전류밀도에서 정전류 충방전실험으로 측정하였다 (그림 7(b)). ZMO400, ZMO500, 그리고 ZMO600 샘플들의 비용량은 각각 199.4, 201.0, 그리고 201.8 mAh g–1로 하소 온도 증가에 따라 비용량이 소폭 증가하였다 (표 4). 앞선 SEM 분석에서 하소 온도 증가는 ZMO 분말의 입자 크기를 증가시켰다(표 1). 활물질 입자 크기의 증가는 전기화학적 활성 면 적(electrochemical active area)을 감소시키므로 일반적으로 비용량을 감소시킨다[54]. 그러나, 높은 하소 온도는 큰 ZMO 입자를 형성함에도 불구하고 비용량은 오히려 소폭 증가하였다. 증가한 비용량은 합성된 ZMO의 전기전도도가 증가하였기 때문으로 생각된다 (그림 7(c)) [55-57]. 왜냐하면, 증가한 전기전도도가 빠른 전자 이동을 가능하게 하여 전극 표면에서 일어나는 산화·환원의 활성화 영역이 더 넓어질 것이기 때문이다[58]. 높은 하소 온도에서 ZMO의 전기전도도가 증가한 원인은 결정구조내 증가한 산소 공공이 shallow donor의 역할을 하였기 때문으로 생각된다[59,60].
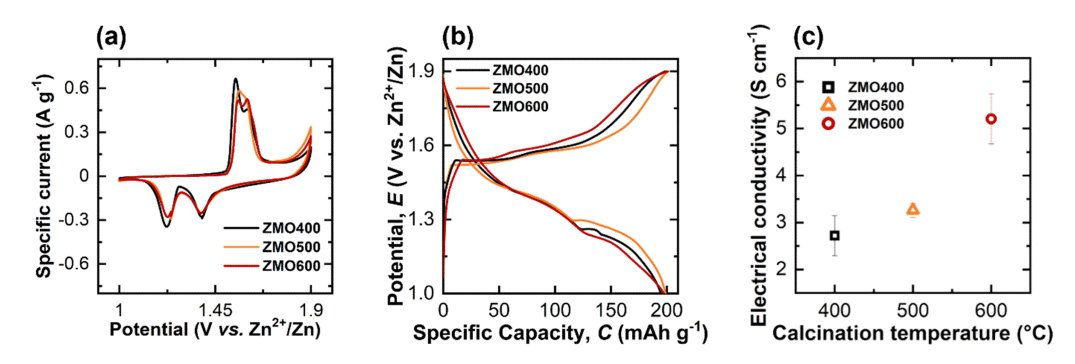
(a) Cyclic voltammograms measured at a scan rate of 0.1 mV s–1, (b) charge/discharge profiles measured at a current density of 0.1 A g–1 for ZMO400, ZMO500, and ZMO600 electrodes, and (c) electrical conductivity measurements of pelletized samples for ZMO400, ZMO500, and ZMO600.

Specific capacity measured at 0.1 A g–1, rate capability in the range of 0.1–2.0 A g–1, and charge transfer resistance (Rct) of the ZMO samples synthesized at different calcination temperatures.
다른 하소 온도에서 합성된 ZMO의 율속 특성 분석을 위하여 다양한 전류밀도(0.1–2.0 A g–1)에서 용량을 측정하였다. 0.1 A g–1에서 측정한 비용량과 2.0 A g–1에서 측정한 비용량을 비교하여 율속 특성을 계산하였다 (그림 8(a), 표 4). ZMO400, ZMO500, 그리고 ZMO600 샘플들의 율속 특성은 각각 46.3, 35.8, 33.2%로 하소 온도가 증가할수록 율속 특성은 감소하였다. 하소 온도 증가에 따 른 율속 특성의 감소는 입자 크기 증가에 따라 길어진 전극 내 Zn2+ 이온의 확산 거리 때문으로 생각된다[61-63]. 또한, ZMO의 입자 크기 증가는 표면적을 감소시켜 전극/전해질 계면에서 전자와 이온의 이동을 저하시키므로 전하 이동 저항(Rct)을 증가시키고 그에 따라 율속 특성도 저하된 것으로 생각된다 (그림 8(b)) [30,62,64,65]. 한편, ZMO 합성시 하소 온도의 증가는 단위 격자 부피를 증가시켜 삽입된 Zn2+의 이동을 용이하게 한다[30,66] (그림 4, 표 2). 또한, 하소 온도 증가에 따른 향상된 전기전도도는 빠른 전자 이동을 가능하게 한다(그림 8(c)). 용이해진 이온의 이동과 빠른 전자 이동 모두 ZMO 전극의 율속 특성 향상에 기여할 수 있다[30,67]. 그럼에도 불구하고 높은 하소 온도에서 합성된 ZMO500, ZMO600 샘플의 낮아진 율속 특성은 입자 크기 증가에 따라 Zn2+ 확산 거리의 증가가 더 큰 영향을 미쳤기 때문이다 (표 1).
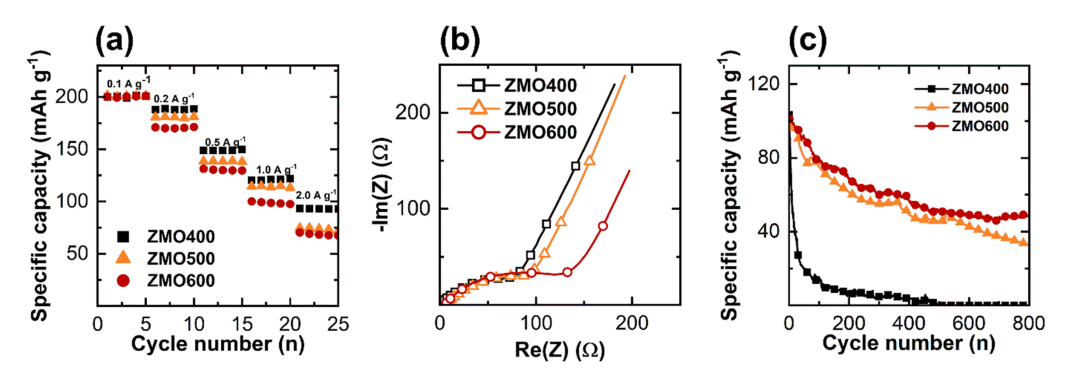
(a) Specific capacity variation at different current density, (b) electrochemical impedance spectra, (c) cycle stability at a current density of 1.0 A g–1 of ZMO samples at different calcination temperature.
ZMO 전극은 충방전시 산화환원 반응을 하는 Mn의 불균등화 반응으로 인해 전해질로 용출되어 용량의 감소를 초래한다[17,68]. 다양한 하소 온도에서 합성된 ZMO전극에서의 수명 특성 분석을 위하여 1.0 A g–1에서 800 cycle 동안 용량 변화를 측정하였다 (그림 8(c), 표 5). 하소 온도 증가에 따른 입자 크기 증가는 수명 특성을 향상시켰다. 특히, 샘플들 중 가장 높은 하소 온도에서 합성된 ZMO600은 48.1%의 향상된 수명 특성을 보였다. 수명 특성이 향상된 이유는 합성된 입자 크기가 증가하여 전극/전해질 계면이 줄어들고, 따라서, 계면에서 발생하는 Mn 용출이 완화되었기 때문으로 생각된다[69]. 앞선 XPS 분석 결과 하소 온도 증가에 따라 용출반응을 일으키는 Mn3+의 비율이 높아졌다. 그럼에도 불구하고 전극 수명의 향상은 전극/전해질 계면의 면적 감소효과가 Mn3+ 비율의 증가 효과보다 더 우세하였기 때문으로 생각된다.
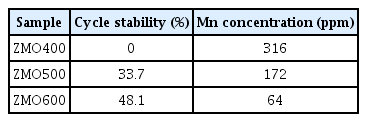
Cycle stability with a current density of 1.0 A g for 800 cycles for ZMO samples at different calcination temperatures. Mn concentration indicates the deposited Mn contents on Zn foil as anode after long–cycle experiment.
높은 하소 온도에서 합성된 ZMO에서 Mn 용출이 완화 되었는지를 분석하기 위하여 Zn 음극에 증착된 Mn 함량을 분석하였다. 불균등화 반응으로 인해 ZMO로부터 용출된 Mn2+는 전해질을 통해 음극으로 이동하여 MnOx 형태로 음극에 전착되기 때문에, 음극에 전착된 Mn 함량은 Mn의 용출량을 나타내기 때문이다[17,25]. 800회 충방전의 수명 특성 분석이 끝난 코인셀을 분해하여 음극으로 사용한 Zn foil 위 Mn의 함량을 측정하였다 (표 5). ZMO600 코인셀의 Zn foil 위 Mn 함량이 64 ppm 으로 가장 낮았다. 따라서, 높은 하소 온도로 인해 큰 입자 크기를 갖는 ZMO600의 경우 전극/전해질 접촉면적 감소로 Mn의 용출이 완화되어 Zn foil 위로 전착된 Mn 의 농도가 감소하였고 이로 인해 가장 우수한 수명 특성을 보였다.
4. 결 론
ZnMn2O4(ZMO)의 수명 특성 향상을 위해서는 Mn의 불균등화 반응을 완화시켜야 한다. 불균등화 반응은 전극/전해질 계면에서 발생하며, ZMO 내 Mn을 전해질로 용출시켜 용량의 감소를 야기하기 때문이다. 따라서, 본 연구는 하소 온도를 높여 ZMO 입자의 성장을 유도함으로써 전극/전해질 표면적을 감소하였다. ZMO400, ZMO500, 그리고 ZMO600의 평균 입자 크기는 각각 12.2, 41.8, 그리고 93.7 nm로 하소 온도가 증가함에 따라 평균 입자 크기가 증가하였다. 높은 하소 온도는 Mn–O 결합을 끊고 격자 구조 내 산소 공공의 함량을 증가시켰으며, 산소 공공이 증가함에 따라 Mn3+의 함량도 증가하였다. 입자 크기 증가에 따라 전기화학적 활성 면적이 감소하였으나 전기전도도의 향상으로 비용량이 소폭 증가하였다. 한편, 입자 크기의 증가와 함께 Zn2+ 이온의 확산 거리가 증가하여 20배 빠른 전류밀도(2.0 A g–1)에서 율속 특성이 46.3(ZMO400), 35.8(ZMO500), 그리고 33.2(ZMO600)%로 감소하였다. 입자 크기 증가로 인해 율속 특성은 감소한 반면, 전극/전해질 접촉 면적이 감소하여 입자 크기가 증가할수록 수명 특성이 향상되었다. 특히, 높은 하소 온도로 인해 가장 입자 크기가 컸던 ZMO600은 800 cycle 동안 48.1%의 가장 우수한 수명 특성을 보였다.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science, ICT & Future Planning) (NRF-2021R1A4A2001658) (NRF-2022R1A2C1009922).